Attachment 'latex_fc28f2d874f1db9e2bb5905927e4a792af392d9d_p1.png'
Download
Attached Files
To refer to attachments on a page, use attachment:filename, as shown below in the list of files. Do NOT use the URL of the [get] link, since this is subject to change and can break easily.- [get | view] (2025-12-10 19:41:22, 1.0 KB) [[attachment:latex_13e1d509a09943b6805b4f75c9bc7ae460ad995b_p1.png]]
- [get | view] (2025-12-10 19:41:21, 0.9 KB) [[attachment:latex_30d98f73695363b8fcdfb42287f2ba2c99f9c1f3_p1.png]]
- [get | view] (2025-12-10 19:41:19, 0.6 KB) [[attachment:latex_33c2ba43d8d752a0963d46b7e9d3a5cfc72a5134_p1.png]]
- [get | view] (2025-12-10 19:41:19, 0.6 KB) [[attachment:latex_4cf781bf6883353e3be1dc5eebb8eb9a3a93db6e_p1.png]]
- [get | view] (2025-12-10 19:41:23, 1.6 KB) [[attachment:latex_4f357442ffc6dea5f1e0700bdd7766b00ae0551a_p1.png]]
- [get | view] (2025-12-10 19:41:20, 0.7 KB) [[attachment:latex_51c6f42ec9881e0ccfa1a2b4fc42858cf538a255_p1.png]]
- [get | view] (2025-12-10 19:41:23, 0.2 KB) [[attachment:latex_585f56f7f6b4da3117e95b0f18f3d5d78e73e92c_p1.png]]
- [get | view] (2025-12-10 19:41:30, 0.6 KB) [[attachment:latex_5bfa5c175479e3ddca67c6ed2ec243be3d662c87_p1.png]]
- [get | view] (2025-12-10 19:41:21, 0.3 KB) [[attachment:latex_5d03221e48131d80d25f421041a336e9d4dd8cf5_p1.png]]
- [get | view] (2025-12-10 19:41:20, 0.7 KB) [[attachment:latex_6085c15f031ee781c43444fc31ae0306ee25167e_p1.png]]
- [get | view] (2025-12-10 19:41:24, 0.8 KB) [[attachment:latex_6705bb30cd88a7c5abce22f463c64d1d79ce5bde_p1.png]]
- [get | view] (2025-12-10 19:41:18, 0.7 KB) [[attachment:latex_6fac61d9e0dc108b60b52791d18bd56afbab1e85_p1.png]]
- [get | view] (2025-12-10 19:41:23, 0.7 KB) [[attachment:latex_7ebd598662864d687d5ced5d1994eea33d085119_p1.png]]
- [get | view] (2025-12-10 19:41:20, 0.8 KB) [[attachment:latex_8d5c7522b6fff0af475a97008f10c8fedfaf31ff_p1.png]]
- [get | view] (2025-12-10 19:41:29, 0.3 KB) [[attachment:latex_92d501fafccd34f831093d6914cf06402628697d_p1.png]]
- [get | view] (2025-12-10 19:41:25, 0.3 KB) [[attachment:latex_94d727df2d08d8e61ee9dd70a2742c0990d7d913_p1.png]]
- [get | view] (2025-12-10 19:41:28, 0.3 KB) [[attachment:latex_978fbeaa4214a20dfbfbfd87d3c6c08af01f9b61_p1.png]]
- [get | view] (2025-12-10 19:41:24, 0.2 KB) [[attachment:latex_986a454dc9f762a8032e30b56dd2f6bff9d4b95a_p1.png]]
- [get | view] (2025-12-10 19:41:19, 0.6 KB) [[attachment:latex_9ba8aed436d0766d9a127be38c8d040802f9797c_p1.png]]
- [get | view] (2025-12-10 19:41:26, 0.3 KB) [[attachment:latex_a1c0232d92060257217c8756d2f63abaf932c109_p1.png]]
- [get | view] (2025-12-10 19:41:21, 0.2 KB) [[attachment:latex_aea06421a467ed22f43134e14bf2c08c43156127_p1.png]]
- [get | view] (2025-12-10 19:41:23, 1.0 KB) [[attachment:latex_b245a091d1d9feab75deb0f03c6d03307ff0f49a_p1.png]]
- [get | view] (2025-12-10 19:41:27, 0.3 KB) [[attachment:latex_b6528bc88fe28f9644663014b211cdb5893cdfc3_p1.png]]
- [get | view] (2025-12-10 19:41:24, 3.3 KB) [[attachment:latex_c6e3351fa63344befd7764275e738c9b30999097_p1.png]]
- [get | view] (2025-12-10 19:41:25, 4.2 KB) [[attachment:latex_cf06112f823cd96b4aa32fae37859961533fbdc6_p1.png]]
- [get | view] (2025-12-10 19:41:22, 0.4 KB) [[attachment:latex_d25cd39a06543ff2b65198e3446a1f772c7247f6_p1.png]]
- [get | view] (2025-12-10 19:41:22, 0.2 KB) [[attachment:latex_d3d89277f224c89b27834b65c828d435241bed66_p1.png]]
- [get | view] (2025-12-10 19:41:21, 0.6 KB) [[attachment:latex_da15fdb7f53cf05a328d1164ee87f0683b243330_p1.png]]
- [get | view] (2025-12-10 19:41:22, 0.3 KB) [[attachment:latex_e17480ab5528e58d8e21651f6c11e403f884026f_p1.png]]
- [get | view] (2025-12-10 19:41:26, 0.3 KB) [[attachment:latex_fc28f2d874f1db9e2bb5905927e4a792af392d9d_p1.png]]
- [get | view] (2025-12-10 19:41:27, 0.4 KB) [[attachment:latex_fde952c5bb0ccdefe9326949427cd70973df3fc8_p1.png]]
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
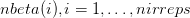{kind=link}
{kind=link}
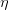{kind=link}
{kind=link}
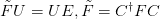{kind=link}
{kind=link}
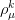{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
You are not allowed to attach a file to this page.